SkriptkommandosDarstellung von Molekülen mit Hilfe von JSmol
Da wie bereits erwähnt, der enorme Funktionsumfang der möglichen Befehle den Rahmen dieses Tutorials sprengen würde, wird hier nur eine kleine Auswahl an Befehlen beschrieben. Eine vollständige Dokumentation findet sich auf chemapps.stolaf.edu/jmol/. Die hier angegebenen Befehle stellen Skripten dar, die jeweils an Steuerelemente gebunden sind, wie sie im vorhergehenden Tutorial besprochen wurden. Entsprechend ihrer Syntax müssen sie in diese eingebaut werden. Die Befehle werden zunächst allgemein erklärt und anschließend auf einer Beispielseite veranschaulicht. Es empfiehlt sich auf der Beispielseite stets den Quelltext aufzurufen, um die exakte Syntax der Befehle zu sehen. Rotation und TranslationDas Skript "rotate Achse Grad;" dreht das gesamte Molekül entsprechend der angegebenen Gradzahl um die angegebene Achse. Als Achse lassen sich X, Y, Z, -X, -Y, -Z angeben. Die Gradzahl kann als Dezimale im Bereich von 0.01 bis 360.00 Grad angegeben werden. Beispielsweise bewirkt das Skript "rotate Y 23.5;" dass das abgebildete Molekül um 23,5 Grad um die Y-Achse entgegen des Uhrzeigersinns gedreht wird. Als Referenz dient das Achsensystem des JSmol Fensters. Auf der Beispielseite können die Skriptbefehle ausgeführt werden. Mit dem Skript "rotate Achse Strecke" wird das Molekül entlang der angegebenen Achse um die angegebene Strecke bewegt. Die Streckenlänge wird in Form einer Dezimalen angegeben, wobei als Einheit Nanometer, Angström oder % gewählt werden kann. "translate X 13.5%;" "translate x -1.0 NM;" "translate y 1.0 ANGSTROMS;" Die Prozentangabe bezieht sich auf die Fenstergröße. Der erste Befehl bewegt das Molekül um 13,5% der Fensterbreite nach rechts. Der zweite Befehl bewegt das Molekül um 1 Nanometer nach links und der dritte Befehl bewegt es um 1 Angström nach unten. Positive Werte verschieben das Molekül auf der X-Achse nach rechts und auf der Y-Achse nach unten. Für negative Werte ist es umgekehrt. Translationen entlang der Z-Achse entsprechen einer Bewegung des Moleküls auf einer Achse senkrecht zur Bildschirmebene und verändern damit die Molekülgröße. Die Molekülgröße kann auch ganz bequem über das Mausrad variiert werden. Die Translationsbefehle werden auf der Beispielseite veranschaulicht. Hinweis: JSmol kann auch die aktuelle Molekülposition anzeigen. Hierzu wählt man im JSmol Menü den Eintrag "Anzeigen" und klickt dann auf "Orientierung". Anschließend öffnet sich ein Fenster, welches die aktuelle Orientierung des Moleküls anzeigt. Dies ist sehr hilfreich, wenn man beispielsweise die Startorientierung eines Moleküls festlegen möchte. Dazu bringt man das Molekül in die gewünschte Position und liest dann die Orientierung aus dem Menü ab. Anschließend können diese Werte in das Ladeskript des Moleküls eingefügt werden.
Das Skript, welches dieses Molekül lädt muss nun durch diese Werte erweitert werden: "load molecule.mol; rotate z -85.21; rotate y 16.22; rotate z 93.16;"
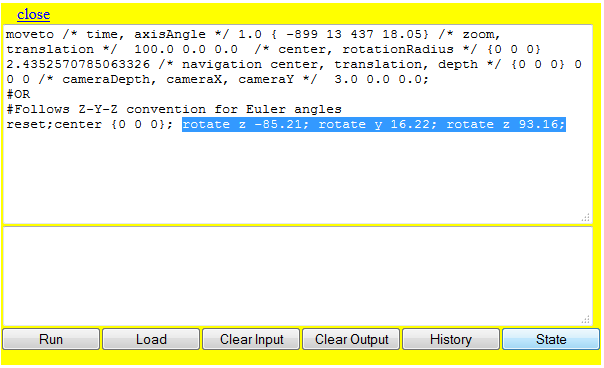
Der "moveto" - BefehlMit Hilfe des "moveto" - Befehls lässt sich das Molekül in eine vorher definierte Position drehen. Wird der Befehl ausgeführt, dann wirkt dies in JSmol so, als ob die Kamera langsam an eine neue Position wandert. Die allgemeine Syntax des Befehls lautet: "moveto Zeit Orientierung" Die Zeit wird in Sekunden angegeben und legt fest wie lang die Bewegung von der Startposition zur Zielposition andauern soll. Die Zielangabe kann auf mehreren Wegen erfolgen. Der einfachste Weg läuft über Schlagworte, die sich auf sechs mögliche Blickrichtungen beziehen:
Der vollständige Befehl, der das Molekül innerhalb von zwei Sekunden von der aktuellen Position zur Frontansicht dreht, lautet: "moveto 2 FRONT;" und kann wie alle folgenden Befehle auf der Beispielseite betrachtet werden. Diese Art der Orientierungsangabe hat den Nachteil, dass sie sich nicht an den Achsen des Moleküls, sondern an den Achsen des JSmol Fensters orientiert. Da bei der Erstellung des Moleküls nicht darauf geachtet wurde, dass dessen Achsen mit den Achsen des Fensters übereinstimmen, wird das Molekül von JSmol auch "schief" geladen. Dies macht auch den "rotate"-Befehl notwendig, sodass das Molekül gerade erscheint. Die zweite Möglichkeit die Zielposition anzugeben ist unabhängig von den Achsen. Dazu dreht man das Molekül zunächst in die gewünschte Position und lässt sich dann über das JSmol-Menü die aktuelle Positionierung des Moleküls anzeigen.
In der ersten Zeile stehen die Orientierungsdaten, die in den "moveto" - Befehl geschrieben werden müssen, um das Molekül wie gewünscht zu drehen. Der Befehl, der eingegeben werden muss lautet dann: "moveto 2 991 -75 110 67.78;" Der "moveto" Befehl kann noch durch Angaben für den Zoom und die Translation erweitert werden. So ist es möglich einen Befehl zu schreiben, der die Ansicht genau auf einen gewissen Punkt im Molekül zentriert. Hierzu positioniert man zunächst das Molekül und ruft dann die Orientierung ab. Die Werte für den Zoom und die Translation können dann einfach abgelesen werden (vgl. Abb. 2). Um sie in den "moveto" Befehl einfügen zu können, müssen die Werte einfach hinter die Werte für die Moleküldrehung geschrieben werden. Der Befehl "moveto 2 991 -75 110 67.78 160;" fügt dem vorherigen Befehl einen Zoom von 160% hinzu. Ebenso können die Werte für die Translation einfach hinter die Werte für den Zoom geschrieben werden: Mit "moveto 2 991 -75 110 67.78 160 -25.2 -17.6;" wird das Molekül zusätzlich noch um die angegebene Strecke -25.2 und -17.6 entlang den Achsen bewegt. Die Angaben beziehen sich wieder prozentual auf die Höhe und die Breite des JSmol Fensters.
AtomauswahlWird ein Molekül von JSmol geladen, so sind standardmäßig alle Atome ausgewählt. Einige Befehle, wie beispielsweise die Anfärbung von bestimmten Atomen, sollen nur auf bestimmte und nicht auf alle Atome wirken. JSmol bietet hier mehrere Möglichkeiten, dies mit Hilfe von Skriptbefehlen zu bewerkstelligen. So können alle Atome des Moleküls ausgewählt werden, nur die Atome eines bestimmten Elementes oder aber auch einzelne Atome nach ihrer Atomnummer, die sie von JSmol zugewiesen bekommen. Standardmäßig ist die Atomauswahl nicht sichtbar, kann aber mit dem Skriptbefehl "selectionHalos ON;" "selectionHalos OFF;"
an- beziehungsweise ausgeschaltet werden. Auf der Beispielseite werden einige Auswahlmöglichkeiten dargestellt, deren Syntax wie folgt lautet:
Beim letzten Befehl auf der Beispielseite, der sich auf die Auswahl der sechs Atome der Peptidbindung bezieht, die sich alle in einer Ebene befinden, wurden die Atome entsprechend ihrer Nummerierung einzeln ausgewählt. Die Atomnummer wird von JSmol angegeben, wenn man den Mauszeiger auf ein Atom bewegt und kurz wartet. Es erscheint dann ein Label, das z.B. C3 # 3 angibt. C3 bedeutet, dass es sich um ein Kohlenstoffatom handelt, #3 gibt die Atomnummer an, mit der das Atom selektiert werden kann. Das Skript, mit dem man mehrere Atomnummern auswählen kann, lautet folgendermaßen: "select atomno=1,atomno=2,...,atomno=n;" Jeder Auswahlbefehl ersetzt dabei den vorhergehenden. Werden erst alle Kohlenstoffatome ausgewählt und dann alle Sauerstoffatome, so sind am Ende nur die Sauerstoffatome selektiert und nicht alle Sauerstoffatome und alle Kohlenstoffatome zusammen.
FärbungStandardmäßig werden Moleküle nach dem "CPK-Farbschema" geladen, das von Coray, Pauling und Kultin entwickelt wurde. Nach diesem Schema werden Kohlenstoffatome schwarz oder grau gefärbt, Wasserstoffatome weiß, Stickstoffatome blau und Sauerstoffatome rot eingefärbt. Möchte man etwas innerhalb eines Moleküls, wie beispielsweise die Peptidbindung auf der Demonstrationsseite, hervorheben, so bietet es sich an, die entsprechenden Atome anders einzufärben. Der Befehl zur Färbung lautet: "color Farbe;" Die Farbe kann dabei auf unterschiedliche Weise spezifiziert werden:
Der "color" Befehl bezieht sich stets auf eine vorher getroffen Atomauswahl. Auf der Beispielseite kann ausprobiert werden, wie sich die Atomfärbung auf die Auswahl bezieht. Mit dem Befehl "color cpk" wird die vorher getroffene Atomauswahl wieder in das CPK-Farbschema überführt. Man spart sich also eine manuelle Auswahl und Färbung aller Atome. Eine Möglichkeit, die der "color" Befehl bietet, ist es, die getroffene Auswahl transparent einzufärben. Mit "color translucent 0.30" werden die selektierten Atome transparent. Die Angabe bezieht sich darauf wie viel Licht hindurch gelassen wird und reicht von 0.1 (1%) bis 1 (100%).
Färbungen beschränken sich nicht nur auf Atome. Bindungen können ebenfalls beliebig eingefärbt werden. Standardmäßig entspricht die Farbe der Bindung jeweils zur Hälfte der Farbe der beiden Atome zwischen denen sie geknüpft wird. Der Befehl "color BONDS Farbe" färbt alle im Molekül vorkommenden Bindungen in der angegebenen Farbe. Um nur eine ganz bestimmte Bindung zu färben, müssen die zwei Atome, zwischen denen die Bindung geknüpft ist selektiert werden und anschlíeßend mit "color BOND Farbe" angefärbt werden.
MoleküldarstellungGängige Moleküldarstellungen sind die Kugel-Stab Darstellung, die Skelett-Darstellung oder die Darstellung als Kalotten-Modell. Wichtig für diese Darstellungen sind Kommandos "spacefill" und "wireframe" "Spacefill" stellt die ausgewählten Atome dabei als schattierte Kugeln dar. Zusätzlich muss der Radius der Kugel angegeben werden, was entweder in Prozent oder in Angström geschehen kann. Die Prozentangabe bezieht sich auf den Van der Waals Radius und darf nur ganze Zahlen enthalten. Die Angabe in Angström darf dagegen auch Angaben mit zwei Dezimalstellen enthalten. Mit "spacefill 10%;" "spacefill 0.1;" werden die ausgewählten Atome als Kugeln mit einem Radius dargestellt, der 10% des van der Waals Radius entspricht, bzw. als Kugeln mit dem Radius 0.1 Angstrom "Wireframe" bezieht sich die dicke der Bindungen zwischen den Atomen und wird in Angstrom angegeben. Der Wert wird als Dezimale angegeben und darf 3.0 nicht überschreiten. Mit "wireframe 0.5;" werden die Bindungen zwischen den ausgewählten Atomen mit einer Dicke von 0.5 Angstrom dargestellt. Der Befehl "wireframe ON;" stellt die Bindungen als Striche dar. Der Parameter OFF schaltet "wireframe" aus "Wireframe" und "Spacefill" müssen miteinander kombiniert werden, um die gewünschten Darstellungen zu generieren.
Manchmal kann es sinnvoll sein, bestimmte Teile des Moleküls auszublenden. Hierzu gibt es zwei Befehle, die in ihren Grundsätzen genau dasselbe bezwecken, sich aber in ihrer Wirkungsweise unterscheiden. Die Befehle lauten "hide selected;" sowie "display selected;" Rückgängig gemacht werden können die Befehle mit "hide none;" und "display all;" Mit "hide selected" wird eine vorher getroffene Auswahl nicht angezeigt. Die Auswahl kann beliebig viele Atome oder Elemente enthalten. Mit "hide BONDS;" werden nur die Bindungen der vorher getroffenen Auswahl nicht mehr angezeigt. Um die Bindungen wieder anzeigen zu können muss "display BONDS;" verwendet werden, da "hide none;" in diesem Fall nicht funktioniert. Der "hide" Befehl muss sich nicht zwangsläufig auf eine Auswahl stützen, er kann auch direkt z.B. auf Elemente angewendet werden. Mit "hide hydrogen;" werden beispielsweise alle Wasserstoffatome des Moleküls ausgeblendet. Der "display" Befehl bewirkt genau das Gegenteil. Eine vorher getroffene Auswahl wird dann angezeigt, alles andere ausgeblendet.
Die Kommandos werden auf der Beispielseite demonstriert.
Darstellung von Atom- und Hybridorbitalen
Erstellen der OrbitaleJSmol ist in der Lage, Atomorbitale (s, p) und Hybridorbitale (sp, sp2, sp3, sp3d und sp3d2) der geladenen Moleküle anzuzeigen. Diese werden nach der LCAO-Methode (linear combination of atomic orbitals) erstellt und entsprechen den Abbildungen, wie sie in Lehrbüchern verwendet werden. Der allgemeine Befehl zur Anzeige von Orbitalen lautet: "lcaocartoon create Orbitalbezeichnung;" In der nachfolgenden Tabelle werden die Orbitalbezeichnungen aufgeführt. Wichtig ist es, dass die Atome, an denen die Orbitale angezeigt werden sollen, vor dem "lcao-Befehl" selektiert werden. Ansonsten kann es sein, dass die Orbitale auch an unerwünschter Stelle angezeigt werden. Die Syntax der Bezeichnungen entspricht der, wie sie in das Skript geschrieben werden müssen.
Um p-Orbitale an einem unhybridisierten Atom anzeigen zu lassen, muss folgender Befehl verwendet werden: "lcaocartoon create \"px\" MOLECULAR;" Dieser Befehl verwendet das Achsensystem von JSmol als Referenz und generiert ein px-Orbital. Neben dem px-Orbital können noch py- und pz-Orbitale dargestellt werden. Hinweis: Ein "-" vor der Orbitalbezeichnung stellt das entsprechende Orbital um 180° gedreht dar.
Orbitalcartoons können auch wieder gelöscht werden, der Befehl lautet: "lcaocartoon delete;" Das Löschen von Orbitalcartoons sollte vor jedem neuen "create" Befehl geschrieben werden, um sicherzustellen, dass die alten Orbitalcartoons entfernt werden bevor neue generiert werden.
Auf der Beispielseite lassen sich die oben aufgeführten Skriptbefehle ausführen. Es wird dringend empfohlen sich den Seitenquelltext aufzurufen, um die komplette Syntax zu betrachten.
Färben der OrbitaldarstellungenStandardmäßig werden die Orbitalcartoons von JSmol eingefärbt. Es ist aber auch möglich die Farben selbständig zu wählen. Der zu verwendende Befehl unterscheidet sich etwas von dem bereits Bekannten. Das allgemeine Skriptkommando lautet "lcaocartoon color [Farbe als Hexadezimale];" Die Farbe selbst wird wie beim bereits bekannten Farb-Befehl in Form einer Hexadezimalen angegeben "[xABCDEF]". Handelt es sich bei dem Orbital um ein p-Orbital, müssen im Skript zum Färben auch zwei Farben angegeben werden, da JSmol beide Orbitallappen einzeln betrachtet. Mit dem Befehl "lcaocartoon color [x000000][xFFFFFF];" wird einer der Orbitallappen schwarz und der andere weiß eingefärbt. Orbitale können wie alle anderen Elemente lichtdurchlässig angezeigt werden, wenn man anstelle der Farbe zu definieren, "translucent" schreibt. Das Maß der Lichtdurchlässigkeit wird wie es bereits bekannt ist, mit Dezimalzahlen angegeben. Befehle, die die Farbe der Orbitale verändern, müssen stets vor einem "lcaocartoon create" Befehl stehen. Sie wirken solange, bis sie durch einen weiteren "lcaocartoon color" Befehl ersetzt werden. Sollen zum Beispiel in einem Wasser-Molekül die freien Orbitale eine andere Farbe erhalten als die bindenden Orbitale, so muss der Befehl folgendermaßen aussehen: "select oxygen;lcaocartoon color [xABCDEF] create \"sp3a\"\sp3b\";lcaocartoon color [xFEDCBA] create \"sp3c\"\"sp3d\"; Die Größe der Orbitallappen kann ebenso variiert werden, indem man folgenden Befehl verwendet: "lcaocartoon scale 1.5;" Der Befehl wirkt ebenso wie der Colorierungs-Befehl auf alle zukünftigen "lcaocartoon create" Befehle, bis er durch einen anderen "lcaocartoon scale" Befehl ersetzt wird.
Darstellung großer Moleküle
Die Darstellung großer Moleküle ist in der Standard Kugel-Stab Ansicht häufig sehr unübersichtlich. JSmol ermöglicht mit Hilfe einiger Skriptkommandos die Darstellung übersichtlicher zu gestalten. Cartoon Darstellung In das JSmol Applet auf der Beispielseite wird beim öffnen ein 3D-Modell des menschlichen Hämoglobins geladen. In der Kugel-Stab Darstellung sind kaum Einzelheiten beziehungsweise die Untereinheiten des Hämoglobins zu erkennen. Mittels des Befehls "cartoon on;" generiert JSmol aus der gegebenen Struktur eine Cartoonabbildung der alpha-Helix und beta-Faltblatt Abschnitte. Der oben genannte Befehl legt jedoch nur die Cartoon-Ansicht über die Kugel-Stab Darstellung, weswegen man diese noch entfernen muss. Das komplette Skript zur Cartoon-Darstellung lautet: "select all;color structure;spacefill 0; wireframe 0; select all;cartoon on;"
Das Skript "color structure" stellt hier keine Farbe dar, sondern definiert analog zu "color CPK" ein Farbschema. JSmol erkennt dann selbständig die einzelnen Sekundärstrukturen und färbt sie entsprechend an. Untereinheiten anfärben Hämoglobin besteht aus vier Untereinheiten, dies wird in der Kugel-Stab Darstellung jedoch nicht deutlich. Nur wenn man mit dem Mauszeiger über die Struktur wandert wird dies anhand der Atomlabels sichtbar. Diese werden im Format [Ala]505:D.CB#3951 angegeben. Dabei gibt der erste Buchstabe, der auf den Doppelpunkt folgt, die Untereinheit (in diesem Fall Untereinheit D) an. Es gibt mehrere Möglichkeiten, die Untereinheiten hervorzuheben. Mit dem Skript "color chain" färbt JSmol selbständig jede Untereinheit in einer anderen Farbe. Da es sich hier aber wieder um ein Farbschema handelt, das von JSmol generiert wird, kann auf die Farbe kein Einfluss genommen werden. Möchte man eigene Farben wählen, so muss man jede Untereinheit manuell selektieren und anschließend mit der gewünschten Farbe einfärben. Der Auswahlbefehl für eine Kette lautet: "select *:A " Dieses Skript selektiert alle Atome der Kette A, welche dann mit dem "color" Befehl beliebig eingefärbt werden können. Auf der Beispielseite können die Befehle ausprobiert werden.
E-Mail: Walter.Wagner ät uni-bayreuth.de, Stand: 27.07.14 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||