Grundlegende Funktionen von JSmolDarstellung von Molekülen mit Hilfe von JSmol
Moleküle die durch das JSmol Applet dargestellt werden, können nach Belieben gedreht und verschoben werden. Weiterhin ist es möglich Bindungslängen sowie Bindungswinkel zwischen verschiedenen Atomen zu messen. Nachfolgend werden die wichtigsten Möglichkeiten aufgeführt.
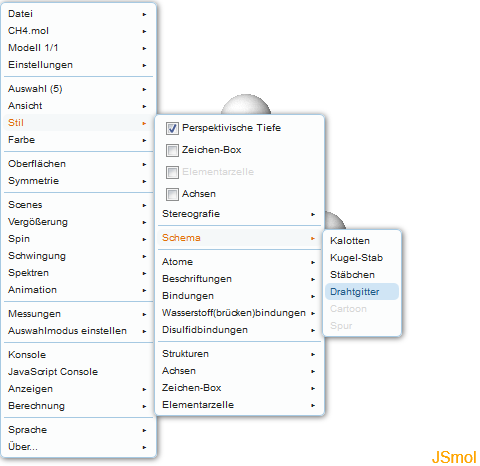
1. MoleküldrehungUm das angezeigte Molekül um die X- und Y-Achse zu drehen, muss man den Mauszeiger in das JSmol Fenster navigieren und ihn bei gedrückter linker Maustaste bewegen. Das Molekül folgt dann den Bewegungen der Maus. Um das Molekül um die Achse senkrecht zur Bildschirmebene zu drehen, muss man zusätzlich die SHIFT-Taste gedrückt halten und die Maus nach links und rechts bewegen. Eine Bewegung nach oben und unten führt bei gedrückter SHIFT-Taste zur Vergrößerung bzw. Verkleinerung des Moleküls. Der Zoom kann auch mit dem Mausrad gesteuert werden. Um das Molekül wieder in die Ausgangsposition zu bringen, kann die Seite mit "F5" einfach erneut geladen werden. 2. MolekülbewegungUm das Molekül in der Darstellungsfläche des Applets zu verschieben, muss man die SHIFT-Taste gedrückt halten und dann zweimal mit der linken Maustaste auf das Molekül klicken, wobei nach dem zweiten Klick die Maustaste gedrückt gehalten werden muss. 3. BindungslängeBindungslängen können gemessen werden, indem man mit der linken Maustaste doppelt auf das Startatom klickt. Wird der Mauszeiger dann in Richtung des Zielatoms bewegt, so zieht dieser eine gestrichelte, rosafarbene Linie hinter sich her. Um die Bindungslänge "einzuloggen", muss man wieder doppelt mit der linken Maustaste auf das Zielatom klicken. Der Abstand wird dann permanent angezeigt, auch wenn das Molekül gedreht wird. Um einen weiteren Abstand zu messen, verfährt man mit zwei anderen Atomen genauso. Eine Aktualisierung der Seite löscht die Messungen wieder. 4. BindungswinkelBindungswinkel misst man prinzipiell genauso wie Bindungslängen. Da jedoch mindestens drei Atome ausgewählt werden müssen, muss man das Startatom doppelt anklicken, das Atom, das den Scheitelpunkt des Winkels darstellt, muss einfach angeklickt werden und das dritte Atom muss man wieder doppelt anklicken. 5. Das JSmol MenüJSmol besitzt ein eigenes Menü, über welches man zahlreiche Einstellungen vornehmen kann. Moleküldarstellung Die Darstellung des Moleküls kann zwischen Kalotten-Modell, Kugel-Stab-Modell, Stäbchen und Drahtgitter gewechselt werden. Dazu öffnet man das JSmol Menü mit einem Rechtsklick und navigiert dann mit dem Mauszeiger über die Menüpunkte "Stil" und "Schema" zur gewünschten Darstellung, die man mit einem Linksklick aktiviert.
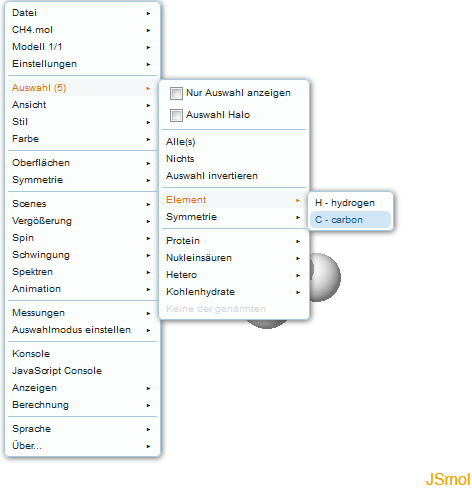
Atomfärbung JSmol bietet auch die Möglichkeit, bestimmte Atome anders einzufärben. Bevor man die Farbe ändern kann, müssen die zu färbenden Atome ausgewählt werden. Dazu öffnet man das JSmol Menü und navigiert über die Menüpunkt "Auswahl" und "Element". Im Menü, welches dann aufklappt, werden alle im Molekül vorhandenen Elemente aufgelistet. Ein Linksklick auf das entsprechende Element wählt alle Atome dieses Elementes aus.
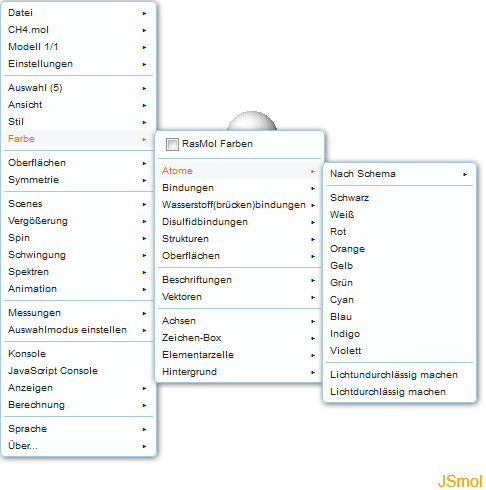
Die Auswahl ist standardmäßig nicht sichtbar, kann aber aktiviert werden, indem man die Checkbox "Auswahl Halo" aktiviert. Die Aktivierung der Checkbox "Nur Auswahl anzeigen" blendet alle nicht ausgewählten Atome aus. Nach der Auswahl einer bestimmten Atomsorte können diese eingefärbt werden. Hierzu muss man über die Menüpunkte "Farbe" und "Atome" navigieren. Das Menü bietet dann mehrere Farben an, die mit einem Linksklick aktiviert werden.
Zur Einfärbung der Bindungen müssen alle Atome ausgewählt werden, da JSmol beispielsweise im Falle einer C-H Bindung die Bindung je zur Hälfte den beiden Atomen zurechnet, zwischen denen sie geknüpft ist. Würde man nur die H-Atome auswählen, würde der Färbe-Befehl nicht funktionieren.
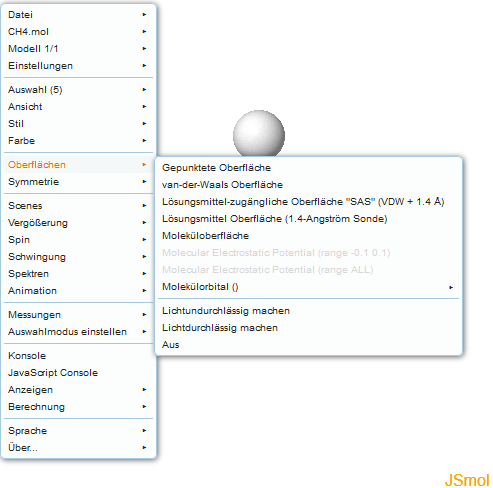
Moleküloberflächen Über den Menüpunkt "Oberflächen" kann beispielsweise die "van-der-Waals-Oberfläche" des Moleküls angezeigt werden.
Einige Menüeinträge sind hier nicht auswählbar oder enthalten keine Menüpunkte. Das liegt daran, dass diese Oberflächen nicht von JSmol selbst berechnet werden können. Die Berechung der MEP Oberfläche muss zum Beispiel mit Hilfe von kommerzieller Software, wie das Programm "Spartan" der Firma Wavefunction, erfolgen. Diese speichern dann diese zusätzlichen Daten mit in der Moleküldatei. Im weiterführenden Turorial wird die MEP-Oberfläche besprochen.
Weitere Funktionen JSmol bietet noch eine Vielzahl von zusätzlichen Funktionen an. Einige davon sind allerdings genauso wie die MEP Darstellung nur dann auswählbar, wenn sie von externen Programmen berechnet und in die Datei mit hineingeschrieben wurden. Ein Beispiel hierfür sind die Darstellungen, die sich hinter dem Menüpunkt "Symmetrie" verbergen. Symmetriedarstellungen und Informationen zur Elementarzelle sind nur den .cif Dateien verfügbar. Einige .cif Dateien sind über die Moleküldatenbank erhältlich.
E-Mail: Walter.Wagner ät uni-bayreuth.de, Stand: 27.07.14 |